Peptide MS-MS spectra data
Proteomics Bulkhttps://www.nature.com/articles/nature19949
Peptide MS/MS spectra data are mass-to-charge ratio (m/z) values and intensities of fragmented peptides ions. MS/MS spectra are analyzed by matching observed fragment ions to theoretical ions from protein databases, enabling accurate peptide and protein identification. Those data are used to determine the amino acid sequence of peptides, quantify proteins, and characterize post-translational modifications in biological samples.
Production systems

nanoHPLC-Exploris 480 coupling
This device offers very high acquisition speed, high resolution (4800000) and allows the use of different acquisition modes(Full-Scan, ddMS2, DIA, PRM). It is used to analyse very complex protein mixtures (identification and quantification).
See more about the technologyAssociated pipelines
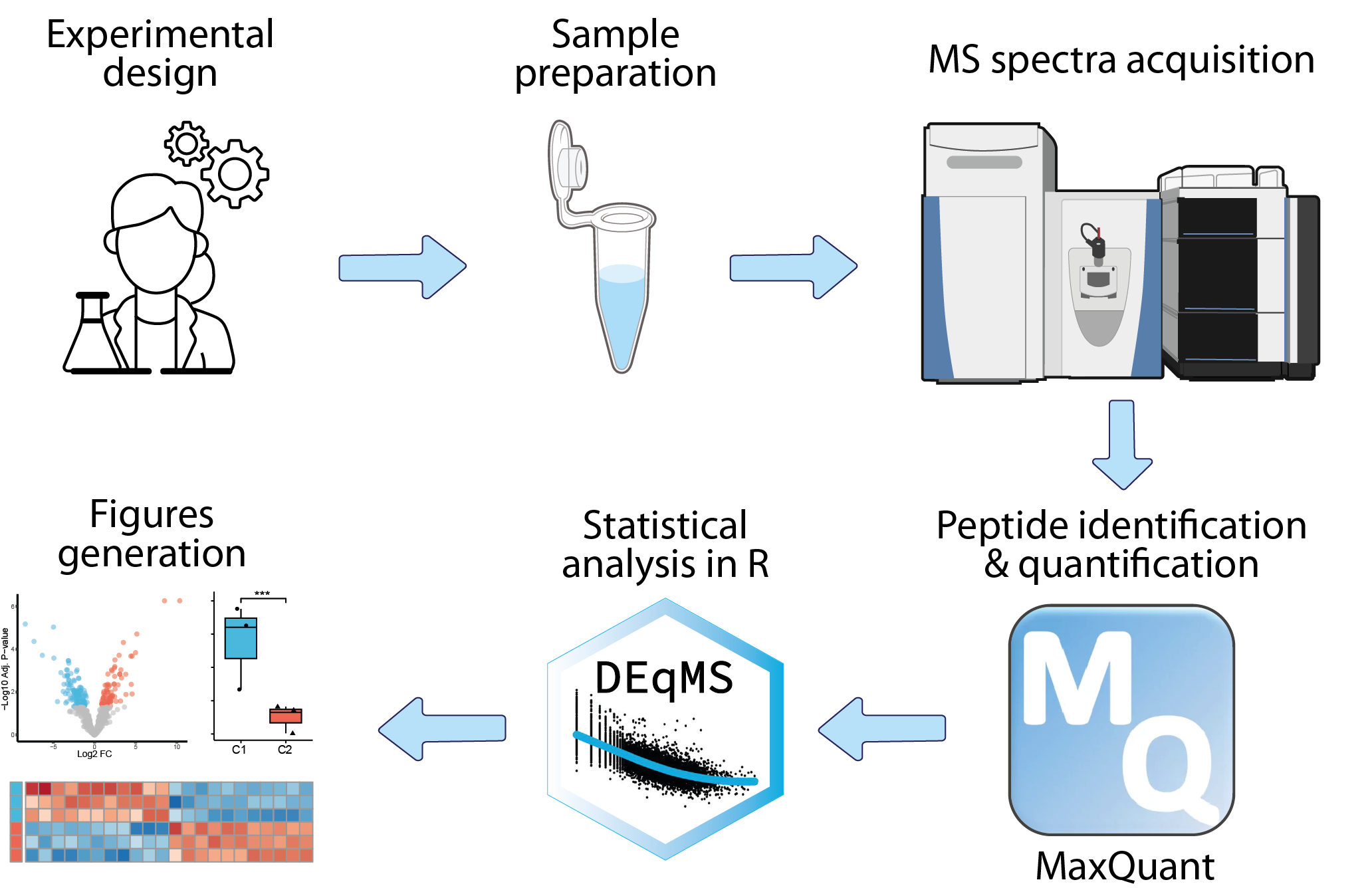
mass-spectrometry-based proteomics
The analysis of mass-spectrometry-based proteomics data begins with the collection of raw mass spectrometry data, which is then preprocessed to remove noise, align spectra, and identify peptide peaks. The next step involves peptide identification, where spectra are matched to known peptide sequences using specialized algorithms. Peptides are subsequently quantified across samples using various strategies, and their associated proteins are identified. This primary analysis is typically conducted by the platform managing the mass spectrometer, utilizing dedicated software such as MaxQuant. The resulting data can then be statistically analyzed within an R framework to identify differentially expressed proteins and enriched biological pathways.
Go to pipeline documentation